Last Updated: September 15, 2022
Introduction to Alkaptonuria
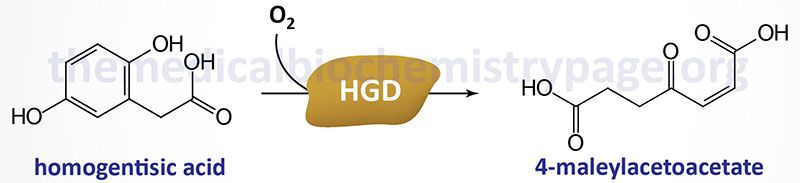
Alkaptonuria is a rare autosomal recessive inherited disorder caused by defects in the gene encoding an enzyme, homogentisic acid oxidase, involved in the catabolism of phenylalanine and tyrosine. As a consequence of this gene defect the catabolism of these two amino acids is inhibited and the intermediate homogentisic acid is excreted in the urine.
If the urine of an individual with alkaptonuria is allowed to stand exposed to the air it will gradually turn dark brown-black as a result of the conversion of homogentisic acid to a melanin-like compound originally called alkapton, hence the derivation of the name of the disorder. The presence of alkali speeds up the conversion process and explains why washing of diapers of afflicted infants makes the stains more pronounced instead of removing them. This striking visual sign of alkaptonuria was key to the initial recognition of the disorder which was first described in detail in 1859. Archibald E. Garrod, in his seminal 1909 publication (Inborn Errors of Metabolism), described the inherited nature of alkaptonuria. In fact, alkaptonuria was not only the first characterized inborn error of metabolism but the first ever disease identified as being inherited.
Alkaptonuria is characterized by homogentisic aciduria, ochronosis (a bluish-black discoloration of tissues) and arthritis. In some alkaptonuria patients there is skin pigmentation in splotches around the mouth. Homogentisic acid accumulation in the heart can lead to valve damage. The exact mechanisms by which alkaptonuria results in ochronosis and arthritis are still not fully understood.
Molecular Biology of Alkaptonuria
Homogentisic acid oxidase is encoded by the HGD (homogentisate 1,2-dioxygenase) gene. The HGD gene is located on chromosome 3q13.33, spans 60 kb and is composed of 16 exons that encode a protein of 445 amino acids.
A number of single nucleotide changes, insertions, deletions, and mutations in introns have been identified in the HGD gene in alkaptonuria patients. In all, mutations have been found in 11 of the 16 exons of the HGD gene.