

Last Updated: March 5, 2024
The Brain as Master Regulator of Feeding Behaviors
The brain, in particular the hypothalamus, plays highly critical roles in the regulation of energy metabolism, nutrient partitioning, and the control of feeding behaviors. The gastrointestinal tract is intimately connected to the actions of the brain in metabolic and appetite control, in a large part, through interactions with the hypothalamic-pituitary axis. These gut-brain interactions occur via the release of gut peptides that exert responses within the brain as well as through neuroendocrine and sensory inputs from the gut. The primary centers in the brain involved in the control of appetite are the hypothalamic-pituitary axis and the brain stem.
The consumption of food initiates a cascade of neuronal and hormonal responses within and by the gastrointestinal system that impact responses in the central nervous system. The brain initiates responses to feeding even before the ingestion of food. The very sight and smell of food stimulates exocrine and endocrine secretions in the gut as well as increasing gut motility. Ingestion of food stimulates mechanoreceptors leading to distension and propulsion to accommodate the food. As the food is propelled through the gut various regions of the intestines secrete numerous hormones that circulate to the brain and impact hypothalamic responses as discussed in the sections below.
The mechanoreceptor responses are transmitted via afferent nerve signals along the vagus nerve to the dorsal vagal complex in the medulla and terminating in the nucleus of the solitary tract (NTS, for the Latin term nucleus tractus solitarii). Projections from the NTS enter the visceral sensory complex of the thalamus which mediates the perception of gastrointestinal fullness and satiety.
Several hormones released from the gut, in response to food intake, exert anorexigenic (appetite suppressing) responses in the brain, particularly in the hypothalamus. These hormones include glucagon-like peptide-1 (GLP-1), cholecystokinin (CCK), peptide tyrosine tyrosine (PYY), pancreatic polypeptide (PP), and oxyntomodulin (OXM or OXY). A single orexigenic (appetite stimulating) hormone, ghrelin, is known to be released by cells of the stomach.
The primary nuclei of the hypothalamus that are involved in feeding behaviors and satiety (the sensation of being full) include the arcuate nucleus of the hypothalamus (ARC, also abbreviated ARH), the dorsomedial hypothalamic nucleus (DMH or DMN), and the ventromedial hypothalamic nucleus (VMH or VMN). The ARC is involved in the control of feeding behavior as well as in the secretion of various pituitary releasing hormones. The DMH is involved in stimulating gastrointestinal activity. The VMH is involved in neural signals that elicit the sensations of satiety.
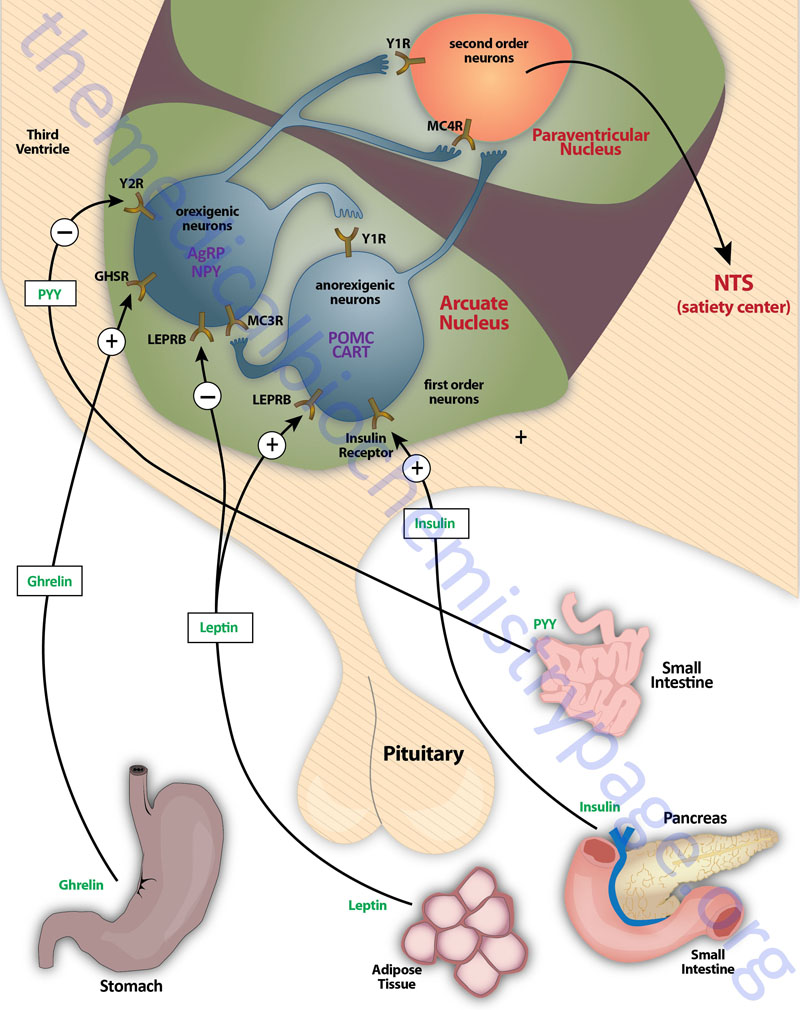
Early experiments involving lesions in the hypothalamus demonstrated that the lateral hypothalamic area (LHA) is responsible for transmitting orexigenic signals (desire for food intake) and loss of this region results in starvation. The medial hypothalamic nuclei (VMH and to a lesser extent the DMH) are responsible for the sensations of satiety and lesions in these regions of the hypothalamus result in hyperphagia (excessive hunger) and obesity.
Appetite is a complex process that results from the integration of multiple signals at the hypothalamus. The hypothalamus receives neural signals, hormonal signals, such as leptin, cholecystokinin (CCK), and ghrelin, and nutrient signals such as glucose, free fatty acids, amino acids, and volatile (short-chain) fatty acids. These effects are processed by a specific sequence of neurotransmitters beginning within domains of the ARC that consist of orexigenic neurons expressing neuropeptide Y (NPY) and Agouti-related peptide (AgRP) as well as domains that consist of anorexigenic neurons expressing pro-opiomelanocortin, POMC (yielding the neurotransmitter α-MSH) and cocaine and amphetamine-regulated transcript (CART).
These ARC neurons are referred to as first order neurons and they release their neurotransmitters to act on second order orexigenic neurons (containing either melanin concentrating hormone, MCH or orexin) or anorexigenic neurons (expressing corticotropin releasing hormone, CRH) to alter feed intake. In addition, satiety signals from the liver and gastrointestinal tract signal through the vagus nerve to the nucleus of the solitary tract (NTS, for the Latin term nucleus tractus solitarii) to cause meal termination, and in combination with the hypothalamus, integrate the various signals to determine the feeding response. The activities of these neuronal pathways are also influenced by numerous factors such as nutrients, fasting, and disease to modify appetite and hence to exert impacts on growth and reproduction.
Metabolic Regulation of Epigenetics in the Brain
The diet has long been understood to play a significant role overall health, life expectancy, and the risk for diseases such as diabetes and cancer. Much of the effect of nutrients in the diet is through alterations that produce long-lasting epigenetic effects. The sensitivity of the epigenome to the diet is due to the fact that constituents of the diet as well as metabolic byproducts of dietary constituents serve as substrates and cofactors for enzymes that covalently modify DNA, RNA, and histone proteins.
Diets that are high in processed foods are known to be high in fats, particularly saturated fats, and sugar. These types of diets have been linked with both mood and neurological disorders, such as depression and memory deficit. Diets high in fats and sugars have also been linked to alterations in behaviors such as learning and memory, sensation of taste, sleep patterns, and importantly overall appetite. Diets high in fats and sugars alter the levels of numerous metabolites involved in modifying the epigenome at both the level of DNA modification and histone modification. High fat and high sugar diets alter the levels of acetyl-CoA through fatty acid β-oxidation and citrate metabolism.
Acetyl-CoA is the substrate for histone acetyltransferases (HAT), enzymes that modify histone functions via acetylation. Increased levels of acetyl-CoA also influence the activity of histone deacetylases, the enzymes responsible for removing acetyl groups. Metabolism of sugars also influences the levels of NAD+ and 2-oxoglutarate (α-ketoglutarate) that in turn effects changes in the activity of histone deacetylases of the sirtuin family (NAD-dependent histone deacetylases) and the 2-oxoglutarate and Fe2+-dependent demethylases that demethylate proteins such as histones as well as methylated DNA.
Many other metabolic intermediates affect the epigenome by serving as substrates for histone modification, most of which are characterized as short-chain lysine acylations. The most well characterized include β-hydroxybutyrylation (Kbhb), lactylation (Klac), crotonylation (Kcr), succinylation (Ksucc), propionylation (Kpr), butyrylation (Kbu), and malonylation (Kmal). Additional lysine acylations have been identified including 2-hydroxyisobutyrylation (Khib), and glutarylation (Kglu).
Diets high in sugars also influence the hexosamine biosynthesis pathway (HBP). The HBP is the pathway responsible for the synthesis of UDP-GlcNAc which is the substrate for protein O-GlcNAcylation. Glucose and glucosamine-6-phosphate drive the rate of UDP-GlcNAc synthesis. In addition to modifying the activity of numerous cytoplasmic and nuclear proteins, such as transcription factors, O-GlcNAcylation of histones represents a major epigenetic modification.
Dietary methionine also influences the epigenome via alterations in the levels of S-adenosylmethionine (SAM or AdoMet). SAM is the primary substrate for DNA methyltransferases (DNMT) and histone methyltransferases (HMT). Since methylation of histones, as well as other proteins, occurs on lysine residues the HMT are also referred to as lysine methyltransferases (KMT).
Studies in animal models have established a clear link between high fat or high sugar diets and cognitive impairment. These effects are associated with changes in DNA methylation as well as the balance of histone acetylation and deacetylation in the brain. Increased methylation of DNA in the promoter regions of several genes associated with memory consolidation in the hippocampus resulted in reduced levels of their expression. One gene in particular, the NAD-dependent histone deacetylase SIRT1 (encoding sirtuin 1) that is hypermethylated in response to diet has been shown to be involved in learning and memory circuitry in the forebrain. Knock down of SIRT1 expression specifically in the brain recapitulates the learning and memory defects observed in mice fed a high fat and high sugar diet. The learning and memory deficits observed in mice fed high fat and high sugar diets is similarly observed in animals fed a high fructose (15%) diet. Results obtained in experimental animals strongly suggest a link between energy-dense diets, DNA methylation, histone acetylation, and cognitive function and have clear implications with the effects of diet on human behaviors such as appetite and the reward pathways elicited by food.
Consumption of high fat and sugar diets has been associated with disrupted feeding behavior leading to weight gain. A major brain region involved in the control of feeding behavior, discussed above, is the hypothalamus, specifically the arcuate nucleus (ARC) of the hypothalamus. Experiments have found that high fat diets lead to increased methylation in the promoter region of the proopiomelanocortin (POMC) gene. One of the products of the POMC gene, the neurotransmitter α-MSH, is a potent anorexigenic neuropeptide. Reduced expression of the POMC gene, due to hypermethylation of the promoter region results in lower levels of α-MSH contributing to increased feeding desire. Although these results were demonstrated in laboratory animals, a positive correlation between POMC promoter hypermethylation and body mass index (BMI) has been established in humans.
Growth Differentiation Factor 15 (GDF15) and Appetite
Introduction to GDF15
Growth differentiation factor 15 (GDF15) is a distant member of the TGF-β superfamily. GDF15 was identified as a cell stress–responsive cytokine and shown to have a primary function as a central regulator of appetite and metabolism. Additionally, GDF15 has been shown to participate in the pathogenesis of a number of disease processes. Despite its official name, GDF15 is not a member of the large GDF family but is instead a divergent member of the glial cell line–derived neurotrophic factor family which itself is a distant subgroup of the TGF-β superfamily.
GDF15 is encoded by the GDF15 gene. The GDF15 gene is located on chromosome 19p13.11 and is composed of 2 exons that encode a proprotein of 308 amino acids. Following synthesis the protein dimerizes through disulfide bonding and is then hydrolyzed at a conserved furin-like cleavage site (RXXR) which generates the functional secreted homodimer of 224 amino acids. The primary proteases responsible for the processing of GDF15 are all members of the proprotein convertase subtilisin/kexin (PCSK) type family with the major proteases being furin (PCSK3), PCSK5, and PCSK6.
GDF15 binds to the receptor identified as glial cell line–derived neurotrophic factor receptor alpha (GFRα)-like, GFRAL. The official name of the gene encoding GFRAL is GDNF family receptor alpha like. The expression of the GFRAL gene is highly localized to two regions of the brain, the area prostema (AP) and nucleus of the solitary tract (NTS: for the Latin term nucleus tractus solitarii). Of the two hindbrain domains, expression of GFRAL is highest in the AP. The AP and the NTS are adjacent hindbrain domains that are both involved in the control of feeding behaviors.
The GFRAL protein is a single transmembrane spanning receptor with no intracellular signaling domain. The binding of GDF15 to GFRAL activates the GFRAL co-receptor, RET, which is a member of the receptor tyrosine kinase family of receptors. The RET receptor initiates a signaling cascade that includes activation of AKT/PKB, ERK1/2, PLCγ, and FOS.
Role of GDF15 in Feeding Behavior
The original observation that GDF15 exerted an effect on controlling appetite and body weight involved the transplantation of human prostate cancer cells into mice. These particular tumor cells overexpressed the GDF15 gene. The mice into which the cells were transplanted developed an anorexia-cachexia syndrome and the degree of body weight loss in these mice was proportional to the serum levels of the human GDF15. The loss of fat mass in these mice was largely due to reduction in food intake. Further demonstration that it was indeed GDF15 inducing these effects came about by blocking GDF15 function with an antibody. In addition, the effects of the tumor cell transplantation could be reproduced by administration of recombinant GDF15. A clinically relevant correlation in humans is that it has been found the in disorders where GDF15 is overexpressed, namely prostate cancer and chronic renal failure, reduction in body mass is correlated to the level of GDF15 in the blood. Subsequent to the original observations with human prostate cancer cell transplantation, the effects of GDF15 were shown to be lost when the GFRAL gene was knocked out in mice.
Numerous experiments have demonstrated the GDF15 plays a physiological significant role in regulating adiposity in humans. The serum level of GDF15 has been shown to change in a diurnal pattern, independent of postprandial increases, which would be expected for a satiety factor. GDF15 reduces reduces the desire for fatty foods and these effects are exerted independent of gastric motility and the vagus nerve. Each of these GDF15 functions indicate that the protein acts as a long-term regulator of energy homeostasis.
Although it is known that GFRAL expression is restricted to the brain, the precise neuronal pathways that mediate the satiety effects of GDF15 are, as yet, incompletely defined. What is known is that GDF15 activation of GFRAL signaling in the AP and NTS triggers activation of neurons in the parabrachial nucleus (PBN) and central amygdala, both of which create a circuit that is known to regulate a desire for food intake.
Expression of GDF15 in adipose tissue increases as the level of adiposity increases. The primary cells responsible for the increased GDF15 are the resident macrophages. The resulting increased serum levels of GDF15 then act through the central
nervous system appetite regulatory circuits leading to a reduced desire to consume food. This process represents an adaptive homeostatic response. Increased expression of GDF15 in the liver also increases as the level of adiposity increases. Evidence in mice indicates that it is the liver-derived GDF15 that is responsible the suppression of appetite, at least in the context of diet-induced obese mice.
The ability of GDF15 to control feeding behavior has ben shown to be cooperative with the effects of leptin. These cooperative effects are the result of interacting neural pathways within the AP and NTS that are activated by leptin binding to its receptor and GDF15 binding to GFRAL. These interconnections between leptin and GDF15, at the level of appetite suppression of induction of body weight reduction, were identified in studies with leptin deficient mice (ob/ob mice). In these mice the efficacy of GDF15 is significantly reduced. Despite the fact that adipose tissue produce more leptin as the amount of fat mass increases, there is a resistance to the effects of this increased leptin within the brain. Nonetheless, the co-administration of leptin to GDF15 treated obese mice results in increased weight loss and reduced adiposity. These observation suggest that pharmacological manipulation of GDF15 in obese individuals is likely to have enhanced benefits by coupling with pharmacologic manipulation of leptin.
Role of GDF15 in Ketogenic Diet-Induced Weight Loss
Ketogenic diets are those that are enriched in fatty acids, adequate protein, and very low levels of carbohydrates. Ketogenic diets were originally developed, and used successfully, in the treatment intractable epilepsy. High fatty acid intake results in increased levels of circulating ketones, primarily β-hydroxybutyrate (BHB). Ketogenic diets are associated with increased energy expenditure, increased insulin sensitivity, reduced serum glucose, and the potential for enhanced longevity.
Recent studies have found that GDF15 plays a major role in the benefits associated with ketogenic diets. Consumption of ketogenic diets is associated with increased production of GDF15 by the liver and a consequent increase in the levels of GDF15 in the blood. This increase in GDF15 production is directly associated with, and required for, the weight loss that accompanies the consumption of a ketogenic diet.
One of the effects of a ketogenic diet is enhanced expression of PPARγ in the liver which in turn enhances the transcription of the GDF15 gene. This effect of PPRAγ appears to be exclusive to the liver since the transcription factor does not enhance GDF15 expression in adipocytes.
Pharmacologic Manipulation of GDF15 Levels
The role of GDF15 in control of appetite along with studies that show increased GDF15 is associated with anorexia and reduced levels are associated with increased food intake indicates that pharmacological intervention in GDF15 levels may be viable in the treatment of obesity and type 2 diabetes. A common drug used to treat the hyperglycemia associated with type 2 diabetes, metformin, has been shown to result in increased serum levels of GDF15. Metformin is also known to cause a reduction in body weight. Recent studies in humans has provided convincing evidence that the reduced appetite (anorexia) and weight loss associated with metformin are indeed mediated by increased plasma levels of GDF15.
The increased plasma GDF15 levels, induced by metformin, are the result of increased expression of the GDF15 gene in the kidneys. This effect of metformin was confirmed in gene knock down studies in mice. Knocking down the level of GDF15 expression, specifically in the the kidneys abolishes the ability of metformin to induce increases in plasma GDF15 levels and also negates the anorexia and weight loss associated with metformin.
Studies are ongoing to ascertain the efficacy of pharmacologic manipulation of GDF15 levels in the treatment of obesity and diabetes. These studies involve the use of recombinant GDF15 or extended half-life GDF15 mimetics. Giving recombinant human GDF15 to diet-induced obese mice results in decreased food intake, body weight, blood glucose, serum insulin,
serum triglyceride, and cholesterol concentrations and improved glucose tolerance in a dose-dependent manner. Extended half-life form of GDF15, which are generated by conjugation to serum albumin or to the immunoglobulin Fc domain, have shown utility in decreasing food intake and weight gain in primates.
Clinical trials of GDF15 mimetics in humans has proven difficult as several companies that began trials have discontinued them for numerous reasons including no demonstration of significant weight loss.
Fructose Effects on Food Intake
As pointed out in the Glycolysis and the Regulation of Blood Glucose page, glucose is the primary fuel used for energy production in the brain. When glucose is metabolized within the hypothalamus, a signaling pathway is initiated that ultimately results in the suppression of food intake. More detailed information on the role of the hypothalamus in the control of food intake can be found in the Gut-Brain Interrelationships page. The principal participants in this signaling cascade include AMPK, acetyl-CoA carboxylase (ACC), and the product of the ACC reaction, malonyl-CoA.
The mechanism by which AMPK and ACC are regulated, ultimately resulting in altered levels of malonyl-CoA are detailed in the Synthesis of Fatty Acids, Triglycerides, and Phospholipids page. Briefly, activation of AMPK results in the phosphorylation of ACC resulting in reduced activity of the latter enzyme. Conversely, when AMPK activity falls the phosphorylation state of ACC falls resulting in increased production of malonyl-CoA. When glucose oxidation is increased in the hypothalamus there is a resultant dephosphorylation and inactivation of AMPK and thereby, activation of ACC. The resultant rise in hypothalamic malonyl-CoA is correlated to reduced expression of several orexigenic peptides (e.g. ghrelin, NPY, and AgRP) concomitant with activated expression of several anorexigenic peptides (e.g. α-MSH and CART). These changes in neuropeptide expression result in suppressed food intake while simultaneously increasing overall energy expenditure.
In contrast to the anorexigenic effect of hypothalamic glucose metabolism, the metabolism of fructose in the brain exerts an orexigenic effect. Although the overall mechanisms by which fructose exerts this orexigenic effect are complex, it is due, in part, to the fact that the brain, like the liver, possesses a unique set of sugar transporters and metabolizing enzymes that enables fructose to bypass the PFK-1 catalyzed step of glycolysis. The PFK-1 catalyzed reaction is the rate-limiting step in glycolysis and is critical in the overall regulation of ATP production and consumption. Since hypothalamic fructose metabolism bypasses this important regulatory step its metabolism rapidly depletes ATP in the hypothalamus. When ATP levels fall there is a concomitant rise in AMP which results in activation of AMPK. Activation of AMPK results in phosphorylation and inhibition of ACC which then results in decreased malonyl-CoA levels in the hypothalamus. Therefore, although glucose and fructose utilize the same signaling pathway to control food intake they act in an inverse manner and have reciprocal effects on the level of hypothalamic malonyl-CoA.
Numerous experiments in animals have implicated malonyl-CoA as a key intermediate in the regulation of feeding behavior and overall energy balance initiated via signaling cascades within the hypothalamus. Initial evidence demonstrating this role of malonyl-CoA was the finding that inhibition of fatty acid synthase (FAS) suppressed food intake. FAS is the primary enzyme in the de novo biosynthesis of fatty acids. The fungal antibiotic, cerulenin, and a related analogue C75, bind to the active site of FAS thereby inhibiting its activity. When these compounds are administered to mice there is a resultant decrease in food intake. Given that inhibition of FAS would be expected to result in an accumulation of its substrate, malonyl-CoA, it was suspected that malonyl-CoA may be a participant in the observed effects. Indeed, intra-cerebrovascular (icv) injection of C75 results in increased levels of hypothalamic malonyl-CoA. Importantly, this effect can be reversed by inhibitors of ACC, the enzyme which synthesizes malonyl-CoA from acetyl- CoA. Further research demonstrated that the anorexigenic effects of FAS inhibitors was due to their ability to rapidly suppress hypothalamic expression of NPY and AgRP, two key orexigenic peptides, while increasing the expression of α-MSH and CART, two key anorexigenic peptides.
Malonyl-CoA levels in the hypothalamus correlate well with nutritional state. Fatty acid synthesis in other tissues, such as the liver and adipose tissue, occurs primarily during energy surplus. Malonyl-CoA, regulates energy metabolism in the liver through two opposing mechanisms. It serves as the substrate for FAS during de novo fatty acid synthesis while at the same time inhibiting mitochondrial fatty acid oxidation through its’ action as an allosteric inhibitor of carnitine palmitoyltransferase 1 (CPT1). Inhibition of CPT1 by malonyl-CoA prevents entry of fatty acids into the mitochondria, thereby inhibiting oxidation. During periods when energy expenditure exceeds intake, such as during fasting, malonyl-CoA levels in the hypothalamus are low. Following food intake there is a rapid rise in hypothalamic malonyl-CoA levels. The changes in hypothalamic malonyl-CoA levels are followed quickly by changes in expression of orexigenic and anorexigenic peptides. In the fasting state NPY and AgRP levels are high, whereas α-MSH and CART levels are low. Upon re-feeding this pattern immediately inverts.
Genetic manipulation of hypothalamic malonyl-CoA levels has also been informative. For example, disruption of FAS gene expression in the hypothalamus results in increased malonyl-CoA levels and decreases in body weight and fat content. Concomitant with disruption of hypothalamic FAS gene expression is an increase in malonyl-CoA concentration. Additionally, the levels of orexigenic peptides decrease while levels of anorexigenic peptides increase leading to suppression of food intake. Conversely, genetic manipulation of the expression of malonyl-CoA decarboxylase (MCD) has the opposite effects to those seen by disruption of FAS expression. MCD functions in opposition to ACC in that it is responsible for the conversion of malonyl-CoA to acetyl-CoA. In fact ACC and MCD are reciprocally regulated to ensure adequate regulation of fatty acid synthesis and fatty acid oxidation.
Overexpression of MCD within the hypothalamus results in increases in both food intake and body weight and also marked increases in body fat content (adiposity). Additionally, when MCD expression is increased within the hypothalamus there is an observed inhibition of the FAS inhibitor (C75)-induced suppression of food intake. These results obtained with forced expression of MCD provide strong evidence that hypothalamic malonyl-CoA acts as an indicator of energy status and participates in the regulation of feeding behavior. In addition, these results indicate that malonyl-CoA, rather than fatty acids, is the effector that regulates energy homeostasis.
Apolipoprotein A-IV and the Control of Feeding Behaviors
Apolipoprotein A-IV (apoA-IV) is synthesized exclusively in the small intestine and the hypothalamus. The APOA4 gene is closely linked to the apoA-I (APOA1), apoA-V (APOA5), and apoC-III (APOC3) encoding genes at chromosomal location, 11q23.3. The APOA4 gene is composed of 3 exons that encode a protein of 46 kDa, derived from a 396 amino acid precursor protein. Utilizing isoelectric focusing it has been determined that two isoforms of apoA-IV, designated A-IV-1 and A-IV-2, can be identified in plasma.
Intestinal synthesis of apoA-IV increases in response to ingestion and absorption of fat and it is subsequently incorporated into chylomicrons and delivered to the circulation via the lymphatic system. Systemic apoA-IV has been shown to have effects in the CNS involving the sensation of satiety.
Intestinal apoA-IV
Following the consumption of fat, the intestinal absorption of the lipid content stimulates the synthesis and secretion of apoA-IV. The increased production of apoA-IV by the small intestine in response to lipid absorption is the result of enhanced transcription of the apoA-IV gene in intestinal enterocytes. The precise signal for this increase in intestinal transcription is the formation and secretion of chylomicrons.
It has been shown that neither digestion, uptake, or the re-esterification of absorbed monoglycerides and fatty acids to form triglyceride is the inducing signal for apoA-IV transcription. This was conclusively demonstrated in experiments showing that the intestinal absorption of only myristic acid or long-chain fatty acids is sufficient to stimulate the lymphatic transport of both chylomicrons and apoA-IV. However, it is still unclear whether different types of triglyceride (those containing either saturated, monounsaturated, or polyunsaturated fatty acids) are equally effective in stimulating the secretion of apoA-IV.
Although it is known that chylomicrons serve as the inducing signal for apoA-IV transcription and secretion, the precise mechanism by which the transcriptional enhancement is effected is currently undetermined. What is known is that an intact vagal innervation from the CNS to the gut is not necessary since vagotomy does not affect intestinal apoA-IV synthesis in response to lipid absorption.
Leptin is a peptide synthesized and secreted by adipocytes whose principal effects result in decreased food intake and increased energy expenditure. The levels of circulating leptin increase in response to the consumption of a high-fat diet and are directly correlated to the amount of fat stored in adipose tissue. The level of apoA-IV transcription has been shown to be reduced within 90 minutes of ingesting a high fat meal and this reduction is a result of increased leptin secretion. Although numerous studies have demonstrated a negative correlation between leptin levels and apoA-IV expression, the mechanism by which this effect is exerted is not fully understood. There are leptin receptors in the gut and, therefore, leptin binding to these receptors could lead to direct effects on intestinal enterocytes.
Alternatively, leptin could exert indirect effects on intestinal cells by increasing fatty acid oxidation through the induction of enzymes that shift fuel metabolism to favor β-oxidation of fatty acids. Given that circulating leptin levels increase as an individual becomes more obese it is likely that leptin is involved in the attenuation of the intestinal apoA-IV response to lipid ingestion. Although the initial response to consumption of a high-fat diet is increased plasma apoA-IV levels, this increase disappears over time. This finding makes it tempting to speculate that the autoregulation of apo AIV in response to chronic high fat feeding is related to the elevation of circulating leptin.
Direct infusion of lipid into the ileum results in increased expression of ileal and jejunal apoA-IV, whereas, infusion of lipid into the duodenum only results in increased jejunal apoA-IV expression. These results strongly suggest that a signal is released by the distal gut during active lipid absorption which is capable of stimulating apoA-IV synthesis in the proximal gut. A strong candidate for this signal is the ileal peptide PYY. To determine if PYY is indeed involved in increased apoA-IV expression experiments were performed in rats involving intravenous injections of physiological doses of PYY.
These experiments showed that PYY infusion does indeed result in significant stimulation of jejunal apoA-IV synthesis and lymphatic transport in fasting animals. Further experiments demonstrated that the stimulation of jejunal apoA-IV synthesis by PYY is the result of effects on translation of the mRNA as opposed to increased transcription of the gene since the levels of the mRNA were unaltered but synthesis of the protein was markedly stimulated. Whereas fat absorption-mediated increases in apoA-IV expression do not require vagal innervation, the responses to PYY do involve the vagal nerve.
Hypothalamic apoA-IV and satiety
Only recently was it determined that both the mRNA and apoA-IV protein are present in the hypothalamus, primarily in the ARC. The presence of apoA-IV in the hypothalamus, a site intimately involved in regulating energy homeostasis, suggests that the effects exerted on appetite by apoA-IV may be due to direct hypothalamic synthesis and secretion. Experiments in rodents, aimed at determining the role of apoA-IV in hypothalamic functions, clearly demonstrated a role for this apolipoprotein in feeding behaviors. Blocking apoA-IV actions by central injection of antibodies to the protein results in increased food consumption, even during the light phase when rodents normally do not eat.
Additional studies have shown that apoA-IV is involved in inhibiting food intake following the ingestion of fat. Infusion of lymph fluid that contains chylomicrons results in markedly suppressed food intake during the first 30 min of administration. However, it is not the lipid content of the chylomicrons that is responsible for the suppression of food intake since infusion of a mixture of triglycerides and phospholipids does not exert the same effect. If apoA-IV is removed from chylomicrons prior to infusion, via the use of specific antibodies, there is no observed effect on food intake. If apoA-IV itself is infused, the level of suppression of food intake is the same as that seen with infusion of fatty lymph fluid containing chylomicrons.
The plasma levels of apoA-IV in humans adapt in response to prolonged consumption of fat. Chronic consumption of a high-fat diet initially results in significantly elevated plasma apoA-IV levels. However, the increased level disappears over time. Conversely, on a low-fat diet, intestinal apoA-IV gene expression is sensitive to fasting and lipid feeding, being low during fasting and high during lipid absorption. Consumption of a high-fat diet results in a slow and progressive reduction in hypothalamic apoA-IV mRNA over time.
The response of hypothalamic apoA-IV gene expression to chronic consumption of a high-fat diet is only partially similar to the response seen in the small intestine. In animals that are chronically fed a high-fat diet there is no observable increase in hypothalamic apoA-IV expression in response to intragastric infusion of lipid following a period of fasting. In contrast, intragastric infusion of lipid into fasted animals that have been consuming normal chow, results in significant stimulation of hypothalamic apoA-IV mRNA levels. These results demonstrate that chronic consumption of a high-fat diet significantly reduces apoA-IV mRNA levels and the response of hypothalamic apoA-IV gene expression to dietary lipids. Therefore, it is highly likely that dysregulation of hypothalamic apoA-IV could contribute to diet-induced obesity.
Metabolites that Control Feeding Behaviors
Intestinal Gluconeogenesis and Feeding Behaviors
Protein-rich diets are known to reduce hunger and subsequent food intake in both humans and experimental animals. In addition, protein-rich, carbohydrate-free diets have been shown to strongly induce the expression of G6Pase, PEPCK-c, and glutaminase in the intestine. In addition, the gut releases glucose to the portal circulation following the intake of a protein-rich, carbohydrate-free diet. The rate of glucose release by the gut can be estimated to provide about 15% to 20% of EGP in protein-fed experimental animals. Significantly, this level of glucose release from the gut is sufficient to account for the level of reduction in food intake observed in protein-fed animals, where an equivalent infusion of glucose into the portal vein of the control animals also decreased food intake and by a comparable value. Although this glucose, derived by intestinal gluconeogenesis, does not increase overall EGP this is because the liver adapts by decreasing its own level of gluconeogenesis while also increasing glycogen storage.
That intestinal gluconeogenesis is indeed crucial in the control of food intake by dietary protein was established with the use mice in which expression of the G6Pase gene was specifically and conditionally abolished in the intestine. When these mice are fed a protein-rich, carbohydrate-free diet they do not exhibit a decrease in their level of food intake such as is seen in control mice on the same diet. The same loss of satiety induction by protein-rich diets or portal glucose infusion is seen in animals whose portal vein afferent nerve connections are chemically or surgically destroyed. Afferent nerves send signals from body locations to the brain. These types of studies demonstrate that portal sensing of intestinal gluconeogenesis is a key mechanism in the satiety effect induced by dietary protein.
Brain areas involved in the control of feeding behaviors include the brain stem and the hypothalamus. For a detailed discussion of the role of the hypothalamus in the control of feeding behaviors visit the Gut-Brain Interrelationships page. In experimental animals fed protein-rich diets or who have had glucose infusions into the portal vein, neuronal activation is observed in several hypothalamic nuclei involved in feeding behavior regulation including the arcuate nucleus (ARC), dorsomedial nucleus (DMN), ventromedial nucleus (VMN), and paraventricular nucleus (PVN). In similar experiments in animals whose gut afferent circuits have been destroyed, there is no increase in neuronal activity following portal vein glucose infusion or consumption of protein-rich diets.
In addition to the effects, on feeding behavior, of intestinal glucose (via gluconeogenesis) delivery to the portal circulation, numerous gut hormones are known to be involved in the control of hunger sensations and do so, in part, via gastrointestinal afferent circuits. The hunger-modulating effects initiated by the release of meal-dependent gut hormones, including cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1), and PYY3-36, are all strongly attenuated by disrupting nerve circuitry between the gastrointestinal and central nervous systems.
Oleoylethanolamide (OEA)
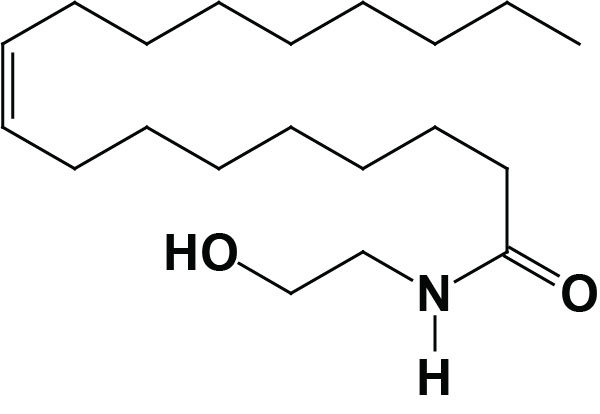
Oleoylethanolamide (OEA; also identified as N-oleoylethanolamine) is a member of the fatty-acid ethanolamide (N-acylethanolamines, NAE) family that includes palmitoylethanolamide (PEA), N-arachidonoylethanolamine (anandamide), N-docosahexaenoyl ethanolamine (synaptamide), and N-stearoylethanolamine. Anandamide was identified as an endogenous ligand (endocannabinoid) for the cannabinoid receptors. Details of the endocannabinoids and the cannabinoid receptors can be found in the Endocannabinoids in Feeding Behavior and Energy Homeostasis page. Synaptamide is an endocannabinoid-like compound produced from the omega-3 PUFA, docosahexaenoic acid, DHA.
Excellent vegetarian and vegan sources of oleic acid are olive oil in which up to 85% of the fatty acid in the triglycerides in olive oil is oleic acid. Other vegetable and nut oils also contain high levels of oleic acid in their triglycerides with canola oil (60%–65%) having the second highest amount compared to olive oil. Pecan oil has 60%–75% oleic acid in its’ triglycerides. Another excellent source of oleic acid (43%) is argan oil (from the argan tree which is abundant in Morocco). In addition to the presence of oleic acid, argan oil has added benefits in that it is high in numerous antioxidant plant phenolic compounds as well as vitamin A and vitamin E. Peanut oil (35%–70%), sunflower oil (20%–80%), and grape seed oil (15%–20%) are also excellent plant sources of oleic acid. Animal fats are also high in oleic acid with lard and tallow containing 44%–47% in the triglyceride component.
OEA Synthesis
Much of the OEA that is absorbed from the intestines is generated by the actions of gut microbiota. However, OEA is also produced by mucosal cells in the proximal small intestine from dietary oleic acid. The pathway of OEA synthesis, like the synthesis of the endocannabinoid anandamide, involves the enzyme N-acyl phosphatidylethanolamine-specific phospholipase D (NAPE-PLD).
Synthesis of OEA occurs on demand within the membrane of the cell by two concerted reactions. The first reaction involves the transfer of a fatty acid residue from the sn-1 position of a phosphatidylcholine (PC) to the free amine of phosphatidylethanolamine (PE). This transfer is catalyzed by a member of the N-acetyltransferase (NAT) family of enzymes. The products of this reaction are known as N-oleoylphosphatidylethanolamines (NOPE) which represent a subclass of the family of lipid molecules called N-acyl-phosphatidylethanolamines (NAPE). At least two activities are termed N-acetyltransferase that are involved in NAPE synthesis (including the synthesis of NOPE), one that is Ca2+-dependent and one that is Ca2+-independent. Although these NAPE synthesis enzymes have been identified and characterized the genes encoding them have not yet been characterized. The Ca2+-dependent NAT is localized to the membrane, whereas the Ca2+-independent NAT is localized to the cytosol indicating that the synthesis of OEA is most likely to be catalyzed by the Ca2+-dependent enzyme.
The second step in synthesis involves cleavage of the NAPE to produce the corresponding fatty acid ethanolamide. This latter reaction is catalyzed by NAPE-specific phospholipase D (PLD) which is encoded by the NAPEPLD gene. The NAPEPLD gene is located on chromosome 7q22.1 and is composed of 8 exons that generates two alternatively spliced mRNAs, both of which encode the same 393 amino acid protein. The NAPEPLD encoded enzyme is unrelated to other members of the PLD family of lipid hydrolases. Once synthesized OEA is eliminated via hydrolysis to oleic acid and ethanolamine. Two enzymes are known to be responsible for OEA hydrolysis. One is known as fatty acid amide hydrolase (FAAH) and the other is PEA-preferring acid amidase (PAA). FAAH is present in the membranes of most mammalian tissues with highest levels observed in brain and liver.
Biological Activities of OEA
OEA has been shown to activate the fatty acid-sensing GPCR identified as GPR119 as well as the non-selective gated cation channel TRPV1 (transient receptor potential vanilloid 1), and to interact with intestinal FAT/CD36 for uptake from the gut. TRPV1 is also known as the capsaicin receptor and is the receptor responsible for the sensation of heat produced by spicy peppers. Evidence suggests that OEA may be the endogenous ligand for GPR119, however, its interaction with FAT/CD36 is required for the satiety response elicited by this bioactive lipid.
OEA is the most potent ligand and likely represents the endogenous ligand for GPR119. The demonstration that OEA is the most active endogenous ligand for GPR119 is of particular interest since previous work has demonstrated that OEA, when administered to laboratory animals, causes a significant reduction in food intake and body weight gain. When intestinal GPR119 is activated by OEA there is clear induction of satiety. However, outside of the intestine the satiety-inducing effects of OEA do not require non-intestinal GPR119 since the effects of systemic OEA administration are still observed in mice in which the gene encoding GPR119 has been knocked out.
GPR119 is, nonetheless, involved in the control of feeding behaviors as demonstrated by studies in GPR119 knock out mice. In response to food deprivation these mice have lower blood glucose and a higher level of body weight reduction when compared to wild-type mice. The response of these mice to food, following food deprivation, is hyperphagia and the time between eating is reduced.
The satiety-inducing effects of OEA involve the activation of the nuclear receptor PPARα resulting in increased expression of fatty acid translocase and modification of feeding behavior and motor activity. The effects are specific to activation of PPARα since data has shown that OEA does not bind PPARγ nor the obligate heterodimer partner of the PPARs, the retinoid X receptors (RXR).
With respect to anorexic effects of fatty acid ethanolamides, OEA is quite specific. This is demonstrated by the use of close structural analogs of OEA which have no effects on feeding behaviors. PEA and anandamide also do not elicit inhibition of feeding. In fact anandamide increase feeding behavior due to activation of the cannabinoid receptor pathway.
Reduced feeding in response to OEA is due solely to an enhanced state of satiety and not to enhanced stress, malaise anxiety or taste aversion. When OEA is administered to rodents prior to the dark period when they prefer to eat, there is a marked increase in feeding latency (delay in onset of feeding) and a decrease in meal frequency. However, there is no effect of OEA on the size of the meal the animals consume. Of significance is the fact that PPARα agonists exert very similar effects on feeding behaviors.
Since OEA is produced in the gut its means of effecting changes in feeding behavior involve engagement of vagal sensory afferent (to the brain) fibers that converge on the nucleus of the solitary tract (NTS) in the brain stem. For more information on the neural circuits functioning between the gut and the brain see the Gut-Brain Interrelationships page. The activation of PPARα by OEA within intestinal enterocytes leads to the activation of the vagal afferent neurons. In animals in which the vagal nerve has been cut OEA fails to reduce feeding behaviors. Further evidence for the role of the vagus nerve in OEA activity is demonstrated by the fact that peripheral administration of OEA results in reduced feeding whereas, intracerebroventricular (ICV) injection does not have an effect. In addition to reducing feeding behavior, OEA administration results in reduced body-weight gain in lean and obese animals. The effects of OEA on weight gain are likely due to increases in energy expenditure since the compound rapidly increases the rate of lipolysis.
There is an self-sustaining activation loop involved in the production of OEA and its effects elicited within the NTS. Activation of neurons within the NTS leads to activation of preganglionic sympathetic neurons within the rostral ventrolateral medulla (RVLM). These preganglionic neurons then activate postganglionic sympathetic neurons that impinge on cells of the submucosal layer and the myenteric plexus. The norepinephrine that is released from the postganglionic sympathetic neurons activates β-adrenergic receptors in these cells leading to increased OEA production. OEA itself exerts effects on the cells of the submucosal layer and the myenteric plexus, likely through binding to GPR119, that result in decreased gastric motility slowing down digestion and activating satiety sensations.
In addition, activation of GPR119 in the pancreas is correlated with enhanced glucose-stimulated insulin secretion (GSIS). Equally important is that activation of GPR119 in the gut results in increased secretion of the incretin hormones GLP-1 and GIP. These observations indicate that GPR119 activation is associated with a dual mechanism of reducing blood glucose: acting directly through pancreatic β-cells to promote GSIS and in the gut via stimulated secretion of the incretins, GLP-1 and GIP, both of which increase insulin release from the pancreas in response to food intake. Currently there are several small molecule agonists of GPR119 in clinical trials being tested for their efficacy in treating the hyperglycemia of type 2 diabetes as well as for their efficacy in treating obesity.
N-Lactoyl-phenylalanine (Lac-Phe)
Exercise stimulates the production of lactate by skeletal muscle. The lactate is primarily released and returned to the liver where it will be used for the synthesis of glucose via gluconeogenesis or taken up by many other tissues for driving TCA cycle activity and ATP synthesis. However, some of the lactate is used in the synthesis of the pseudodipeptide, N-lactoyl-phenylalanine (Lac-Phe).
Lac-Phe is a blood-borne signaling metabolite that suppresses feeding and obesity. As detailed in the Secreted Factors: Tissue “Kines” page, skeletal muscle cells secrete numerous metabolites during and following exercise, not all of which exert inhibitory effects on feeding behaviors. Lac-Phe represents the third highest exercise-induced metabolite secreted from exercising muscle. Other pseudodipeptides, such as Lac-Ile, Lac-Leu, and Lac-Val, have been shown to be equally increased in the blood after exercise but whether they contribute to changes in feeding behavior have, as yet, not been determined.
The biosynthesis of Lac-Phe from lactate and phenylalanine is catalyzed by the non-specific peptidase, carnosine dipeptidase 2 (encoded by the CNDP2 gene). The CDNP2 gene is located on chromosome 18q22.3 and is composed of 15 exons that generate six alternatively spliced mRNAs that collectively encode three distinct protein isoforms. The primary sites of Lac-Phe synthesis are macrophages, monocytes, and other immune and epithelial cells that are localized to a variety of different organs.
In diet-induced obese mice, pharmacologically-mediated increases in Lac-Phe result in reduced food intake without affecting movement or energy expenditure. In contrast, when Lac-Phe is administered to lean mice there is no observed suppression of food intake. Chronic administration of Lac-Phe to diet-induced obese mice decreases adiposity and body weight and improves glucose homeostasis. Conversely, genetic ablation of Lac-Phe biosynthesis in mice increases food intake and obesity following exercise training. In humans there are two known missense mutations in the CNDP2 gene that are associated with increased body mass index.
Plasma levels of Lac-Phe in humans exhibit a sustained increase following exercise, whereas lactate levels will peak at the cession of exercise and will rapidly returned to pre-exercise levels within one hour. The plasma levels of phenylalanine remain unchanged throughout and following exercise. Lac-Phe levels increase and are sustained in various forms of exercise but show the highest levels during and after sprint type exercise. The elevated levels of plasma Lac-Phe, following sprint type exercising, remain for several hours after cessation of the exercise. Endurance training and resistance training also lead to sustained increases in plasma levels of Lac-Phe. In humans, the increases in Lac-Phe during exercise correlate to increases in lean muscle mass and decreases in abdominal subcutaneous and visceral adipose tissue.
The precise target cells for the actions of Lac-Phe have yet to be identified as is the mechanism by which Lac-Phe exerts its suppression of feeding behavior. It is suspected that Lac-Phe binds to a G-protein coupled receptor (GPCR) on target cells, similar to how other metabolites, such as lactate itself, exert their effects.